Identifiers to Enable Pharmaceutical Quality Knowledge Management (PQ KM) – a Progress Report
Version dated: June 2024
Background and Rationale
As pointed out in the 2022 Joint Reflection Paper (JRP), effectively managing changes to pharmaceutical manufacturing processes is challenging, especially to the marketing authorization holders (MAH) submitting the same dossier to multiple regions. Different formats and contents in the dossier are expected due to the country/region specific conventions and legal/regulatory expectations. Such inconsistencies also cause inefficiency amongst regulators and hamper mutual reliance efforts to share knowledge amongst regulators and facilities to be more nimble, agile in regulatory decision making. The envisioned PQ KM system aims at strengthening international collaboration to support global development, manufacture, and supply of pharmaceutical and biological/biotechnological medicinal products.
One key challenge to international collaboration stems from how to identify or confirm that it is the same product under assessment by different national regulatory agencies (NRA) in parallel or at different times. Solving this challenge would allow NRAs to effectively collaborate or share their assessment outcomes and decisions, and eventually make the same product available to the patients in multiple regions sooner. Although some authorities use national or regional identifiers, a common and interoperable limited set of identifiers for manufacturing facilities, pharmaceutical products, substances, marketing applications, and/or marketing application holders is not currently endorsed.
Purpose and Approach:
Recognizing the above challenge, the JRP called for a cross-organization collaboration on defining identifiers. Subject matter experts from the ICMRA/IPRP/ICH/PIC/S member agencies were convened to form a working group. The group is tasked with considering the status of current standards, their implementation, the potential need for new standards and approaches for the development, adoption, operation, governance, and maintenance of a limited set of internationally harmonized identifiers related to pharmaceutical products and the establishments that manufacture them.
In the development phase, the group focused on the key quality information in the eCTD Module 3, and examined the current practices in different NRAs (e.g., EMA, HPRA, PMDA and USFDA) and existing standards and initiatives, such as:
- Key organization information and quality attributes to identify the same product
- The ISO IDMP standards (11615, 11616, and 11238) in identifying medicinal products, pharmaceutical products and substances
- The Substance, Product, Organization and Referential (SPOR) Data Management Services from the EMA
- The Global Substance Registration System (G-SRS) (which provides common unique identifiers (UNII) for all the substances)
- The PIC/S recommendation for unique facility identifiers
The above effort resulted in a proposed identifiers framework, which should help NRAs to quickly confirm the same drug product is under assessment to accelerate or better enable global collaboration and mutual reliance.
Outcome and Discussion:
This framework, as proposed, contains two tiers of identifiers:
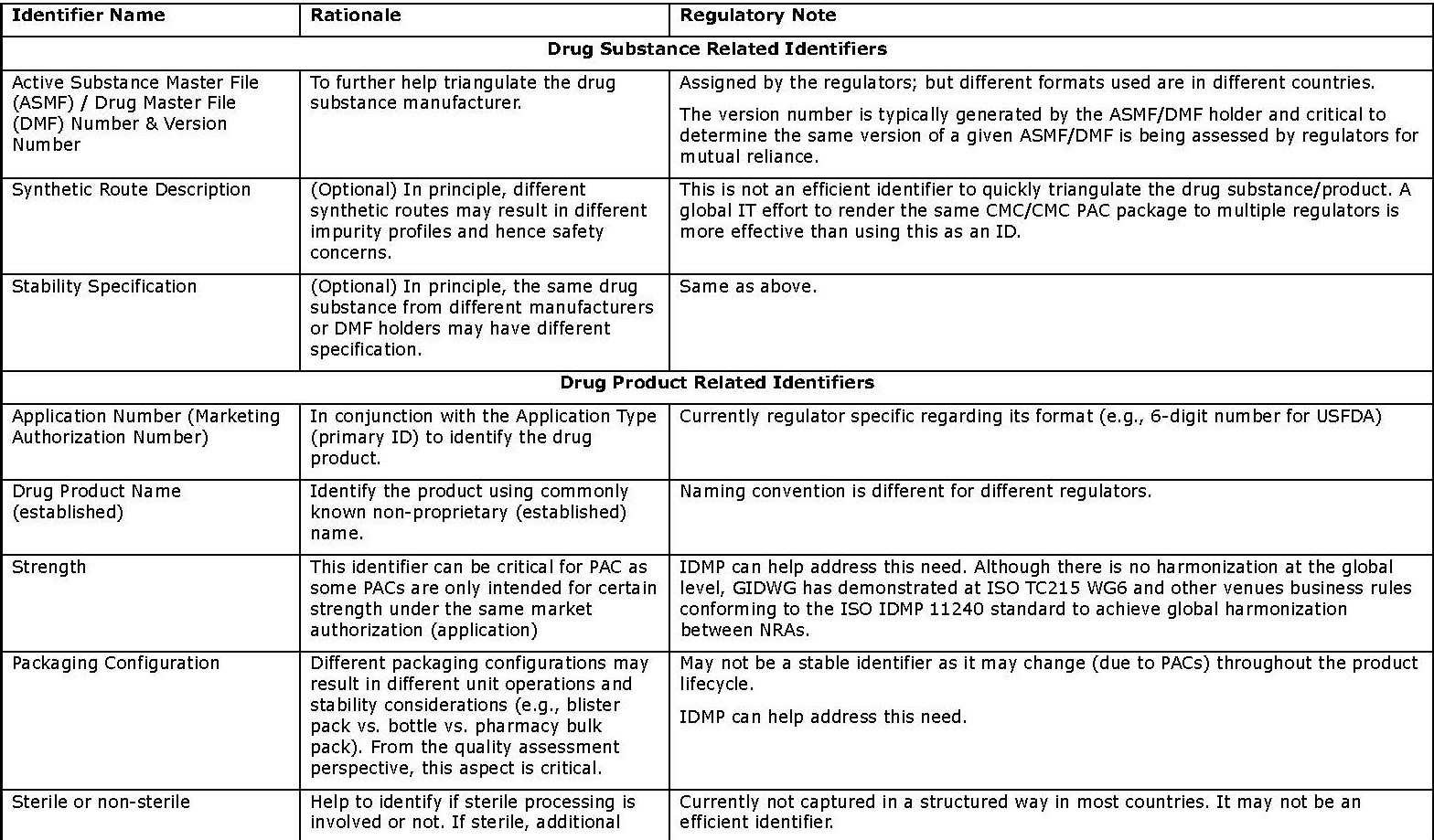
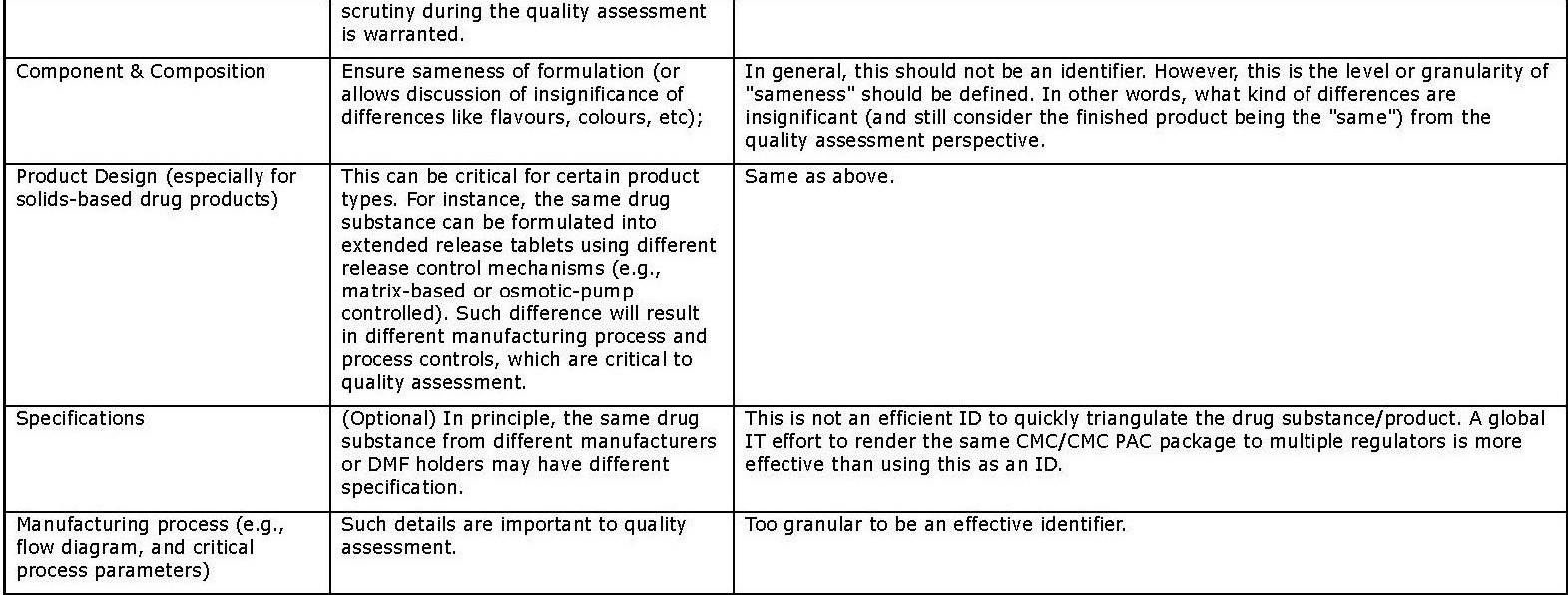
- Primary identifiers (in Appendix 1) are key information about the drug substance and product, and the associated organizations (e.g., MAH and manufacturers). Such information is currently mandatory and submitted in dossiers, typically using well formulated terms or codes in highly structured format. They can be easily located in the submissions (e.g., the cover letter or application form) to serve as effective identifiers. It is estimated that the use of these the primary identifiers should give the NRAs high confidence in determining product “sameness”.
- Secondary identifiers (in Appendix 2) are additional and more granular information about drug substances and products, and associated organizations. Using these identifiers will give a higher level of confidence in determining product “sameness”; however, it may take significantly more effort because they are typically text-based and deeply “buried” in submissions. Therefore, these may be less effective as identifiers.
Regarding the primary identifiers, the work group has a high level of consensus on what the limited set of primary identifiers should be. Also majority of them have been well defined in ISO 11615/11616/11238, which would support their adoption. In fact, these identifiers have been widely and effectively used by different NRAs following their own conventions and practices, including different controlled terms and system generated codes. For instance, FEI and DUNS codes are used by the USFDA to identify manufacturing establishments; however, using such codes is not required by the PMDA. EU/EEA NRAs and EMA use a service called “Organisation Management Service (OMS)” that supports EU regulatory activities and business processes; it stores master data comprising organisation name and location address for organisations such as MAHs, sponsors, regulatory authorities and manufacturers. For application types, different NRAs have different formats due to the respective regional requirements. For dosage form, there is no internationally agreed-upon list of controlled terms. Consequently, the same product can still be labelled differently using the proposed framework, compromising the interoperability of these identifiers across NRAs. Therefore, the successful use of these identifiers for reliance is contingent upon the eventual adoption of internationally recognized and controlled terms or codes.
The group sees the opportunities in both the ongoing and upcoming ICH efforts. The ICH M4Q under revision can clearly specify these identifiers as mandatory for all future submissions and prescribe the desired locations. In addition, the Structured Product Quality Submission (SPQS) effort that will be undertaken in ICH can further harmonize submission content (i.e., data elements and format) and the associated conventions (e.g., the controlled terms and coding strategies). Their implementation and adoption will greatly accelerate the usage of these identifiers; for instance, SPQS can prescribe that all the identifiers are captured in a specific section of a well-structured submission form. To enable such a vision, the Global IDMP working group (GIDWG), formed in 2021, is leading to the establishment of a framework for the global implementation of the ISO IDMP standards and maintenance of global identifiers.
Regarding the next steps for the working group, a deeper dive into the practices and conventions currently used by different NRAs may be insightful to determine the specific differences and potential approaches to harmonize the format/content of these identifiers, to enhance interoperability. The results from this exercise can also serve as a critical reference especially for the upcoming ICH SPQS effort.
Key references:
- The 2022 PQ KMS Joint Reflection Paper
- Considerations on use of unique identifiers to enable reliance through a global regulatory PQ KMS
- EMA PMS ISO IDMP Implementation Guide Chapter 8 Practical example (July 2022)
- ISO 11238: Data elements and structures for unique identification and exchange of regulated information on Substances
- ISO 11239: Data elements and structures for unique identification and exchange of regulated information on pharmaceutical dose forms, units of presentation, routes of administration and packaging
- ISO 11240: Data elements and structures for unique identification and exchange of units of measurement
- ISO 11616: Data elements and structures for unique identification and exchange of regulated pharmaceutical Product information
- ISO 11615: Data elements and structures for unique identification and exchange of regulated medicinal Product information
Appendix 1: Primary Identifiers
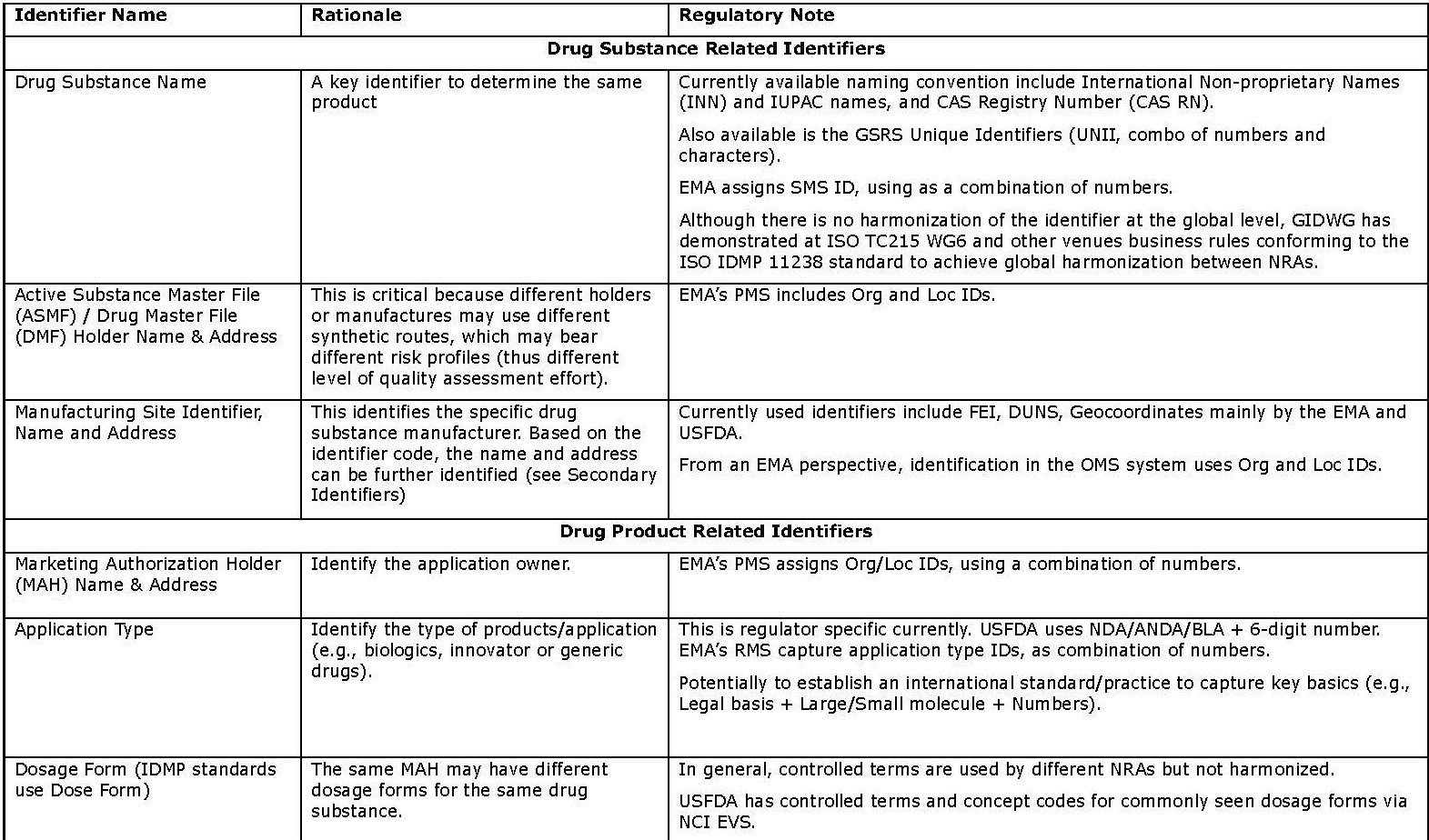
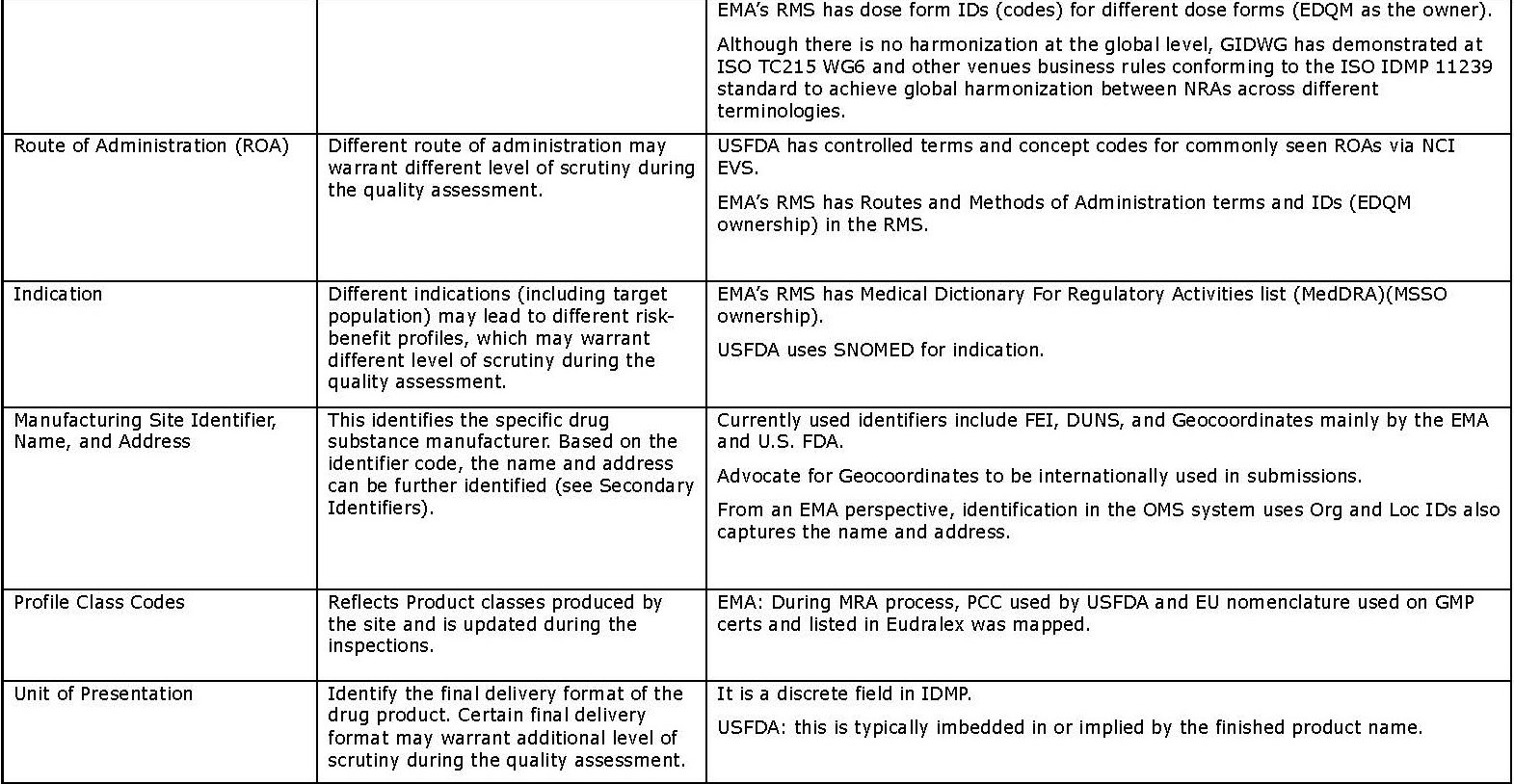
Appendix 2: Secondary Identifiers